

Research
Research area
Our research is focused on the molecular mechanisms of neurodegenerative diseases, most notably FTD (frontotemporal dementia) and ALS (amyotrophic lateral sclerosis). FTD and ALS are currently incurable, and affected patients usually die within a few years of disease onset. Post-mortem brains of FTD and ALS patients show characteristic protein inclusions in the cytosol of neurons and glia cells (Fig. 1). The main components of these pathological inclusions are two RNA-binding proteins, TDP-43 (TAR DNA binding protein of 43 kDa) and FUS (Fused in sarcoma), which usually reside in the cell nucleus and regulate transcription and splicing of numerous target genes.
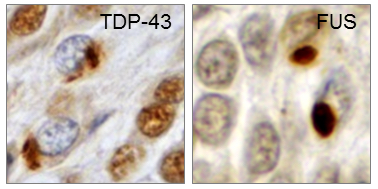
Fig. 1: Neuronal cytoplasmic TDP-43 and FUS inclusions in post-mortem brains of FTD patients. Reproduced from Dormann & Haass, Trends Neurosci 2011
We have previously shown that proper nuclear import of FUS is crucial for neuronal health (Dormann et al. 2010; Dormann & Haass 2011). This is best exemplified by our finding that ALS-causing point mutations in the FUS gene disrupt the nuclear localization signal of FUS and thus impair nuclear import of FUS mediated by the nuclear import factor Transportin/Karyopherin ß2 (Fig. 2). Moreover, we found that nuclear import of FUS is regulated by a post-translational modification, arginine methylation of FUS, and that this modification is defective in FTD patients with FUS inclusions (Dormann et al. 2012; Suarez-Calvet et al., 2016). Currently, we study how nucleocytoplasmic transport of FUS and TDP-43 are regulated, e.g. by post-translational modifications or RNA-binding
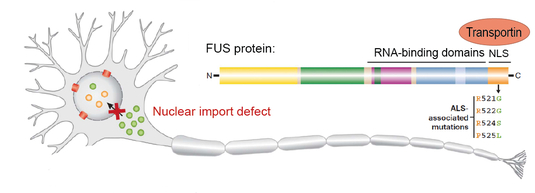
Fig. 2: Defective nuclear import of FUS causes ALS. ALS-associated point mutations in the NLS of FUS weaken Transportin binding and thus impair nuclear import of FUS.
Furthermore, our previous work has shown that stress granules (SGs) may act as condensation points, where TDP-43 and FUS start to aggregate once they exceed a certain critical concentration. This conclusion is based on the observation that elevated cytosolic levels of TDP-43 and FUS (e.g. due to NLS mutations) strongly favor the proteins’ localization in SGs, and that TDP-43 and FUS aggregates in post-mortem brains of ALS and FTD patients contain SG marker proteins (Dormann et al., EMBO Journal 2010; Bentmann et al., J. Biol. Chem. 2012). This led us to propose the “two-hit model” (Dormann & Haass, Trends in Neurosciences 2011; Bentmann et al., FEBS Journal 2013), which postulates that (i) nuclear import defects and (ii) SG formation are important pathological hits leading to TDP-43 or FUS aggregation (Fig. 3).
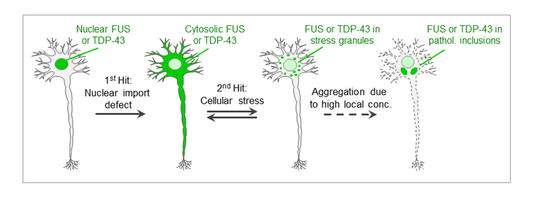
Fig. 3: “Two-hit model”. Nuclear import defects (1st hit) and cellular stress (2nd hit) cause clustering of cytosolic TDP-43 or FUS in stress granules (SGs), triggering their aggregation due to high local protein concentrations.
Recent work from our group suggests that aberrant liquid liquid phase separation (LLPS) plays an important role in FUS-associated neurodegeneration. In LLPS, a homogenous solution of molecules separates into a dense phase that is enriched in certain molecules and a phase that is depleted of these molecules. FUS contains amino acid sequences that drive such LLPS, leading to the coalescence of FUS molecules in liquid-like protein droplets (Fig. 4). Once formed, such liquid droplets can harden into gel- or solid-like structures. Thus, LLPS can potentially be pathological and cause detrimental protein aggregation. We have recently shown that aberrant LLPS of FUS is prevented by arg-methylation as well as by binding to its nuclear import receptor Transportin/Karyopherin ß2 (Hofweber, Hutten et al., 2018). Hence, loss of FUS arg-methylation (as seen in FTD patients) and impaired Transportin binding (as seen in ALS patients, Fig. 1) both promote LLPS of FUS (Fig. 4).
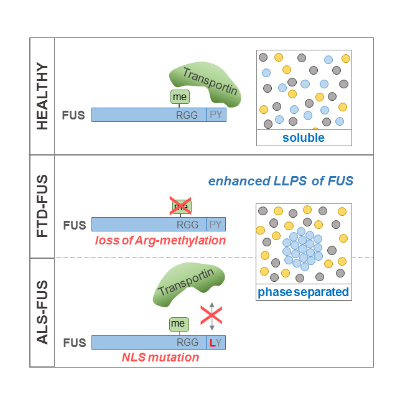
Fig. 4: Two distinct mechanisms suppress liquid-liquid phase separation
(LLPS) of FUS and are compromised in FTD and ALS.
Research projects
To gain insight into the molecular mechanisms that underlie TDP-43 and FUS mislocalization and aggregation and subsequent neurodegeneration, we study the intracellular transport pathways and aggregation behavior of TDP-43 and FUS as well as their interactions with RNAs and other RNP granule components using in vitro and cell culture models (cell lines, primary neurons) and a combination of cell biological methods, molecular biology and protein biochemistry.
Currently, we are interested in the following research questions:
(1) Nuclear transport defects in ALS/FTD: By which mechanisms are TDP-43 and FUS exported from the nucleus to the cytoplasm? Which additional factors regulate nuclear import and export of TDP-43 and FUS? Are nuclear transport components or regulatory factors defective in ALS/FTD patients? How does the most frequent genetic cause of ALS and FTD, a repeat expansion in the C9orf72 gene, affect the nucleocytoplasmic transport machinery?
(2) Post-translational modifications of RNA-binding proteins: How does arginine methylation regulate protein-protein interactions, RNA binding and the functions of FUS and other methylated RNA-binding proteins? How does loss of FUS arginine methylation arise in FTD-FUS patients? Which other post-translational modifications regulate the physiological and pathological roles of FUS and TDP-43?
(3) LLPS and RNP granules: How do FUS mutations alter the composition of cytosolic FUS RNP granules and cytosolic mRNA processing functions of FUS? Which factors suppress or enhance pathological LLPS of FUS and TDP-43 in RNP granules? What is the physiological role of LLPS of FUS and TDP-43?
Complete publication list:
https://www.ncbi.nlm.nih.gov/pubmed?term=(Dormann D[Author] AND Munich[All Fields]) OR (Schmid D[Author] AND Munz C[Author])
Selected publications
Bourgeois B*, Hutten S*, Gottschalk B, Hofweber M, Richter G, Sternat J, Abou-Ajram C, Göbl C, Leitinger G, Graier WF, Dormann D#, Madl T# (2020). Nonclassical nuclear localization signals mediate nuclear import of CIRBP. Proc Natl Acad Sci U S A.117(15):8503-8514. * equal contribution; # = co-corresponding authors
Alberti S, Dormann D (2019). Liquid-Liquid Phase Separation in Disease. Annu Rev Genet. 53:171-194.
Ukmar-Godec T, Hutten S, Grieshop MP, Rezaei-Ghaleh N, Cima-Omori MS, Biernat J, Mandelkow E, Söding J, Dormann D, Zweckstetter M (2019). Lysine/RNA-interactions drive and regulate biomolecular condensation.
Nat Commun. 10(1):2909.
Hofweber M, Dormann D (2019). Friend or foe - post-translational modifications as regulators of phase separation and RNP granule dynamics. J. Biol. Chem 294(18):7137-7150.
Hofweber M*, Hutten S*, Bourgeois B, Spreitzer E, Niedner-Boblenz A, Schifferer M, Ruepp MD, Simons M, Niessing D, Madl T, Dormann D (2018). Phase separation of FUS is suppressed by its nuclear import receptor and arginine methylation. Cell, 173(3):706-719.e13
Ederle H, Funk C, Abou-Ajram C, Hutten S, Funk EBE, Kehlenbach R, Bailer S, Dormann D (2018). Nuclear egress of TDP-43 and FUS occurs independently of Exportin-1/CRM1, Scientific Reports, 8(1):7084.
Ederle H, Dormann D (2017). TDP-43 and FUS – en route from the nucleus to the cytoplasm.
FEBS Letters, 591(11):1489-1507
Khosravi B, Hartmann H, May S, Möhl C, Ederle H, Michaelsen M, Schludi MH, Dormann D, Edbauer D (2017). Cytoplasmic poly-GA aggregates impair nuclear import of TDP-43 in C9orf72 ALS/FTLD. Hum Mol Genet. 26(4):790-800
Bowden HA, Dormann D (2016). Altered mRNP granule dynamics in FTLD pathogenesis. J Neurochem. 138 Suppl 1:112-33.
Suárez-Calvet M, Neumann M, Arzberger T, Abou-Ajram C, Funk E, Hartmann H, Edbauer D, Kremmer E, Göbl C, Resch M, Bourgeois B, Madl T, Reber S, Jutzi D, Ruepp MD, Mackenzie IR, Ansorge O, Dormann D*, Haass C* (2016). Monomethylated and unmethylated FUS exhibit increased binding to Transportin and distinguish FTLD-FUS from ALS-FUS. Acta Neuropathol. 131(4):587-604.
Bentmann E, Haass C, Dormann D (2013). Stress granules in neurodegeneration – lessons learnt from FUS and TDP-43. FEBS J. 280(18):4348-70
Dormann D, Madl T, Valori C, Bentmann E, Tahirovic S, Abou-Ajram C, Kremmer E, Ansorge O, Mackenzie IR, Neumann M, Haass C (2012). Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. EMBO J. 31(22):4258-75
Bentmann E, Neumann M, Tahirovic S, Rodde R, Dormann D*, Haass C* (2012). Requirements for stress granule recruitment of Fused in sarcoma (FUS) and TAR DNA-binding protein of 43 kDa (TDP-43). J Biol Chem. 287(27):23079-94
Dormann D, Haass C (2011). TDP-43 and FUS – A nuclear affair. Trends Neurosci. 34(7) pp. 339-348
Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than M, Mackenzie I, Capell A, Schmid B, Neumann M, Haass C. (2010) ALS-associated FUS mutations disrupt Transportin-mediated nuclear transport. EMBO J. 29(16):2841-57
Dormann D, Capell A, Carlson AM, Shankaran SS, Rodde R, Neumann M, Kremmer E, Matsuwaki T, Yamanouchi K, Nishihara M, Haass C. (2009) Proteolytic processing of TAR DNA Binding Protein-43 by caspases produces C-terminal fragments with disease defining properties independent of progranulin. J Neurochem. 110(3):1082-94
Schmid D, Pypaert M, Münz C. (2007) MHC class II antigen loading compartments continuously receive input from autophagosomes. Immunity 26(1):79-92
Paludan C*, Schmid D*, Landthaler M, Vockerodt M, Kube D, Tuschl T, Münz C. (2005) Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 307(5709):593-6
Note: Since 2007 Schmid D = Dormann D
Funding
We are supported by grants of the Deutsche Forschungsgemeinschaft (DFG) (Emmy Noether grant DO-1804/1-1, DFG grant DO-1804/3-1 and DFG grant DO-1804/4-1 within the priority programme SPP2191) and by the Munich Cluster for Systems Neurology (SyNergy, EXC 2145). We are also supported by a research grant of the Fritz Thyssen Foundation.
We also received Seed Funding by the LMU Junior Researcher Fund within the framework of LMUexcellent and by the Heinz Maier-Leibnitz Award of the DFG (DO-1804/2-1).




